Фенилкетонурия (ФКУ) у детей
Фенилкетонурия (ФКУ) у детей — генетическая болезнь, которая характеризуется нарушениями обмена фенилаланина и бывает у 1 из 8000–15 000 новорожденных. Форм фенилкетонурии (ФКУ) всего 4, но существует 400 разных мутаций и метаболические фенотипы заболевания.
Фенилкетонурия — наследственная аминоацидопатия, при которой снижается интеллект ребенка, и возникает неврологический дефицит.
Фенилкетонурия I (классическая или тяжелая) – это аутосомно-рецессивное заболевание, которое возникает вследствие мутации гена фенилаланингидроксилазы. В основе заболевания лежит нехватка фенилаланин-4-гидроксилазы которая обеспечивает превращение фенилаланина в тирозин, результатом чего становится накопление фенилаланина и его метаболитов в тканях и физиологических жидкостях организма ребенка.
Отдельную группу представляю атипичные варианты фенилкетонурии. При них симптомы очень похожи на таковые при классическом варианте заболевания. Но нет положительных продвижений по показателям развития ребенка, даже если проводить нужную диетотерапию. Такие варианты объясняются нехваткой дегидроптеринредуктазы, тетрагидроптерина, гуанозин-5-трифосфатциклогидролазы, 6-пирувоилтетрагидроптеринсинтазы и пр.
Фенилкетонурия II (атипичная) — аутосомно-рецессивная болезнь, при которой генный дефект находится в коротком плече хромосомы 4. Характеризуется она нехваткой дегидроптеринредуктазы, что приводит к нарушению восстановления активной формы тетрагидробиоптерина, а в спинномозговой жидкости и сыворотке крови снижается уровень фолатов. Результат таких изменений – метаболические блоки в механизмах превращения фенилаланина в тирозин. Заболевание было выявлено еще в конце 20-го века.
Фенилкетонурия III (атипичная) — аутосомно-рецессивная болезнь, которая вызвана недостаточностью 6-пирувоилтетрагидроптеринсинтазы. Он принимает участие в организме в процессе создания тетрагидробиоптерина из дигидронеоптеринтрифосфата, что было открыто в конце 20-го столетия. Нарушения сходы с таковыми при выше описанной (второй) форме.
Примаптеринурия — атипичная фенилкетонурия у детей с легкой гиперфенилаланинемией, у которых присутствует в больших количества в моче примаптерин и часть его производных, а в спинномозговой жидкости нормальная концентрация нейромедиаторных метаболитов.
Материнская ФКУ – болезнь, при которой снижается уровень интеллекта (вплоть до умственной отсталости) среди потомства женщин, которые больны фенилкетонурией и не сидели на специальной идете, когда были совершеннолетними.
Есть предположения, что при материнской ФКУ нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита. Было проведено исследование в 2008 году Кочем и его командой. У младенца, рожденного от матери с ФКУ, при аутопсии головного было найдено некоторое количество патологических изменений: вентикуломегалия, низкий вес мозга, задержка миелинизации (признаков астроцитоза не наблюдалось), гипоплазия белого вещества.
В некоторых странах СНГ применяется условная классификация рассматриваемого заболевания по уровню содержания в сыворотке крови фенилаланина:
Фенилкетонурия у детей и ее лечение
Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина. Встречается с частотой 1 на 8000–15 000 новорожденных. Выделяют четыре формы ФКУ; существует свыше 400 различных мутаций и несколько метаболических фенотип
.jpg) Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина. Встречается с частотой 1 на 8000–15 000 новорожденных. Выделяют четыре формы ФКУ; существует свыше 400 различных мутаций и несколько метаболических фенотипов ФКУ [1].
Фенилкетонурия (ФКУ) — генетическое заболевание, характеризующееся нарушениями обмена фенилаланина. Встречается с частотой 1 на 8000–15 000 новорожденных. Выделяют четыре формы ФКУ; существует свыше 400 различных мутаций и несколько метаболических фенотипов ФКУ [1].
Определение, патогенез, классификация
Фенилкетонурия — наследственная аминоацидопатия, связанная с нарушением метаболизма фенилаланина, в результате мутационной блокады ферментов приводящая к стойкой хронической интоксикации и поражению ЦНС c выраженным снижением интеллекта и неврологическим дефицитом [1, 2].
Основное значение в патогенезе классической ФКУ имеет неспособность фенилаланингидроксилазы перерабатывать фенилаланин до тирозина. В результате в организме накапливается фенилаланин и продукты его аномального обмена (фенилпировиноградная, фенилуксусная, фенилмолочная кислоты) [1–3].
В числе других патогенетических факторов рассматриваются нарушения аминокислотного транспорта через гематоэнцефалический барьер, нарушения церебрального пула аминокислот с последующим нарушением синтеза протеолипидных белков, нарушения миелинизации, низкие уровни нейротрансмиттеров (серотонин и др.) [1–4].
Фенилкетонурия I (классическая или тяжелая) — аутосомно-рецессивное заболевание, вызванное мутацией гена фенилаланингидроксилазы (длинное плечо хромосомы 12); выявлены 12 различных гаплотипов, из которых около 90% ФКУ ассоциировано с четырьмя гаплотипами. Наиболее частые мутации в гене фенилаланингидроксилазы: R408W, R261Q, IVS10 nt 546, Y414C. В основе болезни — дефицит фенилаланин-4-гидроксилазы, обеспечивающей конверсию фенилаланина в тирозин, что приводит к накоплению в тканях и физиологических жидкостях фенилаланина и его метаболитов [1–4].
Особую группу составляют атипичные варианты ФКУ, при которых клиническая картина напоминает классическую форму болезни, но по показателям развития, несмотря на проведение диетотерапии, не отмечается положительной динамики. Эти варианты ФКУ связаны с дефицитом тетрагидроптерина, дегидроптеринредуктазы, 6-пирувоилтетрагидроптеринсинтазы, гуанозин-5-трифосфатциклогидролазы и т. д. [1–4].
Фенилкетонурия II (атипичная) — аутосомно-рецессивное заболевание, при котором генный дефект локализуется в коротком плече хромосомы 4 (участок 4р15.3), характеризующееся недостаточностью дегидроптеринредуктазы, приводящей к нарушению восстановления активной формы тетрагидробиоптерина (кофактор в гидроксилировании фенилаланина, тирозина и триптофана) в сочетании со снижением в сыворотке крови и спинномозговой жидкости фолатов. Результатом являются метаболические блоки в механизмах превращения фенилаланина в тирозин, а также предшественников нейромедиаторов катехоламинового и серотонинового рядов (L-дофа, 5-окситриптофан). Болезнь описана в 1974 г. [1–4].
Фенилкетонурия III (атипичная) — аутосомно-рецессивное заболевание, связанное с недостаточностью 6-пирувоилтетрагидроптеринсинтазы, участвующей в процессе синтеза тетрагидробиоптерина из дигидронеоптеринтрифосфата (описано в 1978 г.). Дефицит тетрагидробиоптерина приводит к расстройствам, сходным с нарушениями при ФКУ II [1–4].
Примаптеринурия — атипичная ФКУ у детей с легкой гиперфенилаланинемией, у которых в моче в больших количествах присутствует примаптерин и некоторые его производные при наличии нормальной концентрации в спинномозговой жидкости нейромедиаторных метаболитов (гомованилиновой и 5-оксииндолуксусной кислот). Энзиматический дефект пока не выявлен [1–4].
Материнская ФКУ — заболевание, сопровождающееся снижением уровня интеллекта (до умственной отсталости) среди потомства женщин, страдающих ФКУ и не получающих специализированную диету в совершеннолетнем возрасте. Патогенез материнской ФКУ детально не изучен, но предполагается ведущая роль хронической интоксикации плода фенилаланином и продуктами его аномального метаболизма [1–4].
R. Koch и соавт. (2008) при аутопсии головного мозга младенца, у матери которого отмечалась ФКУ (без адекватного контроля за уровнем фенилаланина в крови), обнаружили ряд патологических изменений: низкий вес мозга, вентикуломегалию, гипоплазию белого вещества и задержку миелинизации (без признаков астроцитоза); хронических изменений в сером веществе головного мозга не было обнаружено. Предполагается, что нарушения в развитии белого вещества мозга ответственны за формирование неврологического дефицита при материнской ФКУ [5].
В практических целях в медико-генетических центрах РФ используется условная классификация ФКУ, основанная на уровнях содержания фенилаланина в сыворотке крови: классическая (тяжелая или типичная) — уровень фенилаланина выше 20 мг% (1200 мкмоль/л); средняя — 10,1–20 мг% (600–1200 мкмоль/л), а также уровень фенилаланина 8,1–10 мг%, если он устойчив на фоне физиологической нормы потребления белка в рационе питания; легкая (гиперфенилаланинемия, не требующая лечения) — уровень фенилаланина до 8 мг% (480 мкмоль/л) [2].
Клинические проявления и диагностика
При рождении дети с ФКУ I выглядят здоровыми, хотя чаще имеется специфический хабитус (светлые волосы, голубые глаза, суховатая кожа). При отсутствии своевременного выявления и лечения болезни в течение первых двух месяцев жизни у них появляется частая и интенсивная рвота и повышенная раздражительность. Между 4 и 9 месяцами становится очевидным выраженное отставание в психомоторном развитии [1–4].
Пациентов отличает специфический («мышиный») запах кожных покровов. Выраженные неврологические нарушения у них редки, но характерны черты гиперактивности и расстройств аутистического спектра. При отсутствии своевременного лечения уровень IQ составляет
В. М. Студеникин, доктор медицинских наук, профессор
Т. Э. Боровик, доктор медицинских наук, профессор
Т. В. Бушуева, кандидат медицинских наук
НЦЗД РАМН, Москва
Что такое фенилкетонурия? Причины возникновения, диагностику и методы лечения разберем в статье доктора Алексенцева Е. С., врача УЗИ со стажем в 12 лет.

Определение болезни. Причины заболевания
Фенилкетонурия (ФКУ) — генетическое заболевание, в основе которого лежит врождённое нарушение метаболизма аминокислот, характеризующееся повышенным содержанием фенилаланина в крови. Это аутосомно-рецессивная патология, т. е. ребёнок может унаследовать данное заболевание только в том случае, если оба родителя являются носителями дефектной версии гена.
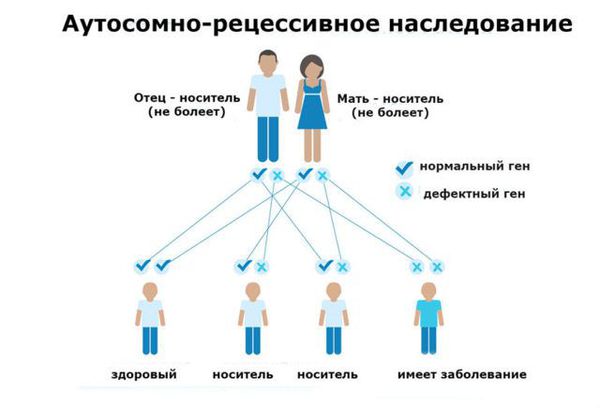
Распространённость фенилкетонурии
Факторы риска фенилкетонурии
Основной фактор риска фенилкетонурии — это наличие у обоих родителей дефекта в гене PAH (Phenylalanine hydroxylase gene). Заболевание развивается, если оба родителя передают ребёнку копию повреждённого гена.
Симптомы фенилкетонурии
Как проявляется фенилкетонурия у новорождённых
Чем дольше ребёнок с фенилкетонурией не получает специфического лечения, тем быстрее развивается умственная отсталость и необратимые нарушения развития. Кроме того, для детей с фенилкетонурией характерны следующие признаки [4] :
Патогенез фенилкетонурии
Фенилкетонурия как самостоятельное заболевание было открыто норвежским врачом Иваром Асбьёрном Фёллингом ещё в 1934 году. Несмотря на это, вопрос о патогенезе долгое время оставался открытым.
Фенилаланин — это незаменимая аминокислота, которая участвует с синтезе белков. Незаменимая она потому, что организм не может самостоятельно её синтезировать, фенилаланин можно получить исключительно из пищи (мясных и рыбных продуктов, творога, сыра, яйц, орехов, хлебобулочных изделий, круп) или с помощью протеолиза — процесса гидролиза белков с помощью ферментов-протеаз.
У пациентов, страдающих фенилкетонурией, из-за дефекта гена и недостатка фермента фенилаланин-гидроксилазы происходит увеличение в плазме крови концентрации фенилаланина (более 1200 мкмоль/л при норме 0-120 мкмоль/л ) и его метаболитов. Одновременно с этим снижается уровень тирозина и его производных (дофамина, адреналина, норадреналина и меланина). Такое состояние оказывает выраженное нейротоксическое действие на структуры мозга. Если пациент с фенилкетонурией не получает или не соблюдает лечение, то у него отмечаются повреждения мозолистого тела, полосатого тела, изменения коры и гипомиелинизация (снижение содержания миелина, образующего оболочку нервных волокон, в различных структурах оболочек мозга). Эти изменения могут привести к снижению интеллектуального развития и нейродегенерации — прогрессирующей гибели нервных клеток. Поэтому пациенты с фенилкетонурией более восприимчивы к нарушениям, связанным с дефицитом дофамина в головном мозге, таким как паркинсонизм.
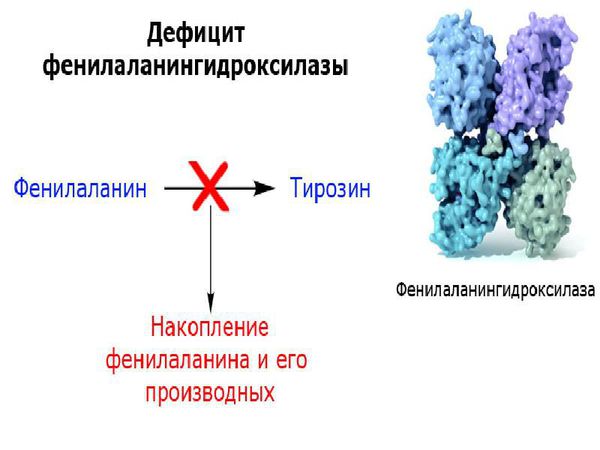
Хотя патофизиологические механизмы повреждения головного мозга у пациентов с фенилкетонурией ещё не совсем понятны, существует множество свидетельств метаболических изменений, которые включают:
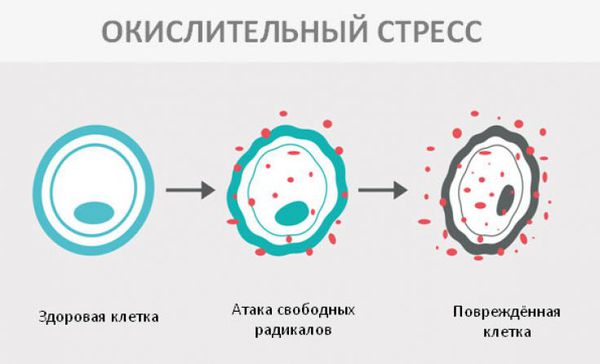
Окислительный стресс — это повреждение клеток активными формами кислорода, которые представляют собой молекулы с повышенной реактивностью из-за наличия неспаренного электрона на внешнем электронном уровне. Активные формы кислорода образуются в клетках постоянно, но в норме их уровень настолько низкий, что организм самостоятельно их нейтрализует с помощью антиоксидантной системы. Окислительный стресс происходит в том случае, когда активных форм кислорода образуется слишком много и антиоксиданты не могут полностью их инактивировать. Такой дисбаланс может вызвать окислительное повреждение белков, липидов или ДНК.
Классификация и стадии развития фенилкетонурии
Пожизненная диетотерапия ассоциирована с нарушением роста, снижением минеральной плотностей костей и дефицитом питательных веществ, что требует постоянного контроля у профильных специалистов.
Диагностика фенилкетонурии
Развитие медицины привело к созданию тандемной масс-спектрометрии для быстрого определения концентраций аминокислот в небольших объёмах крови или плазмы. Данный метод даёт более низкую частоту ложноположительных результатов, измеряя уровни фенилаланина и тирозина в исследуемых образцах.
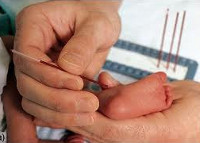
Для неонатального скрининга медицинским персоналом с помощью скарификатора осуществляется забор крови из пятки новорождённого строго через 3 часа после кормления. Полученные образцы крови помещаются на специальные фильтровальные бумажные тест-бланки и отправляются в лабораторию.
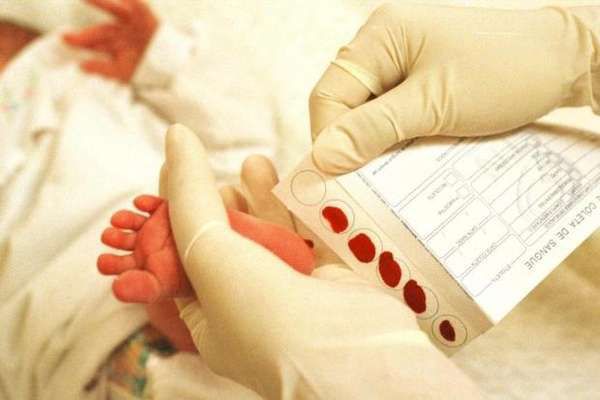
Стоит отметить, что у некоторых детей, особенно у рождённых раньше срока, может наблюдаться незрелость ферментных систем, участвующих в метаболизме аминокислот. Это приводит к кратковременному повышению фенилаланина и положительному результату при скрининге [12] Около 2 % всех случаев повышенного уровня фенилаланина в крови, выявленного при скрининге новорождённых, связаны с нарушением метаболизма кофермента BH4, который участвует в преобразовании фенилаланина. Это подчёркивает важность проведения дифференциальной диагностики при всех выявленных уровнях гиперфенилаланинемии. Фенилкеторурию необходимо дифференцировать с такими заболеваниями, как транзиторная гиперфенилаланинемия недоношенных, наследственная доброкачественная гиперфенилаланинемия, тирозинемия, галактоземия.
Лечение фенилкетонурии
Контроль уровня фенилаланина в крови
Мнение медицинского сообщества относительно начала лечения пациентов с концентрациями фенилаланина 360-600 мкмоль/л достаточно противоречивы. Costello и соавторы [18] проводили исследование, в котором пациенты были разделены на три группы:
Общее потребление белка должно обеспечивать безопасные уровни потребностей данного макронутриента с дополнительной дотацией 40 % L-аминокислот (20 % L-аминокислот необходимы для компенсации потребностей в незаменимой аминокислоте и еще 20 % L-аминокислот используются для контроля фенилаланина в крови).
При введении неадекватных дозировок L-аминокислот они ограничивают синтез белка. Белковый обмен, который в норме складывается из процессов анаболизма (синтеза) и катаболизма (распада) белков, смещается в сторону катаболизма. При данном процессе фенилаланин остается неиспользованным для синтеза белка, и его концентрация в крови будет расти.
При фенилкетонурии необходимо избегать продуктов, богатых белком (мясо, рыба, яйца, обычный хлеб, большинство сыров, орехи и семена), а также продуктов, содержащих аспартам (подсластитель, который используется при изготовлении некоторых газированных изделий и конфет). Пища с низким содержанием белка при диетотерапии данного заболевания должна содержать 50 мг или менее фенилаланина на 100 г сухого продукта. Фрукты и овощи, содержащие менее 75 мг фенилаланина на 100 г пищевого продукта, также могут быть включены в рацион. При употреблении таких овощей, как картофель, брокколи, цветная капуста, брюссельская капуста, нужно учитывать, что даже их небольшое количество в рационе обеспечивает организм 50 мг фенилаланина, поэтому их потребление не должно быть бесконтрольным. Для удобства родителей и ребёнка следует пользоваться «пищевым светофором», где все продукты разделены на группы, разрешённые (ограниченно или неограниченно) и запрещённые к употреблению.
Когда ребёнок маленький, соблюдение диеты не является проблемой для семьи, так как родители контролируют потребление продуктов. В младенческом возрасте предлагается использовать специализированные смеси без добавок фенилаланина, которые дополняются либо грудным молоком, либо стандартной семью для перекрытия суточной потребности в фенилаланине.
Дети более старшего возраста продолжают не только пить специальный безфенилаланиновый продукт, который способен обеспечить потребности в белках и калориях, но и получают дополнительное количество разрешённых продуктов (овощи и фрукты, мёд, животные и растительные масла, зефир, пастила, варенье), необходимых для создания пищевого разнообразия.
По мере взросления ребёнка соблюдение диеты становится всё труднее, так как дети с фенилкетонурией, в отличие от своих сверстников, значительно ограничены в выборе продуктов, что часто приводит к скачкам концентрации фенилаланина у подростков [20] [21] Долгосрочное поддержание диеты необходимо, поскольку пациенты после периода нарушений диеты намного труднее возвращаются к прежнему режиму питания.
Прогноз. Профилактика
Если пациенты с фенилкетонурией получают и соблюдают лечение с самого раннего возраста, то их качество и продолжительность жизни ничем не отличается от их здоровых сверстников.
Пациенты, не получающие или не соблюдающие лечение, часто имеют инвалидность и низкий уровень качества жизни. Кроме того, несоблюдение диеты и отсутствие контроля фенилаланина в организме часто приводит к снижению продуктивности и внимания, нарушению поведения (особенно при уровне аминокислоты свыше 360 мкмоль/л).
Адекватное наблюдение за концентрацией фенилаланина в пределах допустимых показателей достаточно эффективно в профилактике большинства нарушений центральной нервной системы. Большинство людей демонстрируют нормальное общее развитие, легко справляются с образовательными стандартами, ведут самостоятельную жизнь и получают работу, будучи взрослыми.
При соблюдении пациентом режимов диеты и дополнительной дотации минеральных веществ не отмечается увеличения риска таких осложнений, как остеопороз или частые переломы при отсутствии других заболеваний костно-мышечной системы и соединительной ткани.
Профилактика фенилкетонурии
Если у будущих родителей или их близких родственников выявлена фенилкетонурия, то при планировании беременности рекомендуется проконсультироваться с генетиком и пройти обследование. Так можно определить риск рождения ребёнка с фенилкетонурией.
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
МКБ-10
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.